

赵杨与万里研究组揭示动植物保守的TIR酶活磷酸化刹车机制——渗透胁迫下平衡植物免疫与生长的关键
干旱、盐胁迫以及低温等非生物逆境导致植物受到渗透胁迫,严重威胁粮食安全。植物在胁迫响应中可能存在‘有害应答’,即免疫信号过度激活(如NLR蛋白通过未知途径被诱导),其不仅拮抗ABA介导的胁迫耐受通路,还会导致生长停滞,严重威胁非生物逆境下的植物生存。如何限制免疫信号超激活的“有害应答”,是渗透胁迫下植物生长维持的关键。
2025年6月9日,中国科学院分子植物科学卓越创新中心、植物高效碳汇重点实验室(中国科学院)赵杨研究组在国际学术期刊Nature Plants在线发表题为“TIR immune signalling is blocked by phosphorylation to maintain plant growth”的研究论文。该研究揭示了一种动植物保守的免疫‘刹车机制’:通过磷酸化TIR结构域抑制其NADase活性,从而精准调控免疫信号强度,使植物在胁迫下兼顾防御与生长。
赵杨研究组之前鉴定到渗透早期信号元件BON1,发现其通过调控Ca2+信号协调渗透应答,并负调控NLR蛋白介导的免疫信号(Chen et al.,2020, Curr Biol)。渗透胁迫下,bon1突变体的免疫信号过度激活,从而严重抑制其生长。通过抑制子筛选,团队发现SNC1(编码一种含TIR结构域的NLR蛋白)的多个显性失活突变(insensitive to osmotic stress,ios)可完全恢复bon1突变体的生长,证实SNC1是植物维持渗透胁迫下生长的核心因子。TIR结构域通过切割NAD+启动防御反应,但过度激活会导致细胞死亡和生长停滞。因此,动态调节TIR信号的“开”与“关”,是植物实现“防御生长两不误”的关键。
赵杨研究组近期研究发现,CPK3/4/6/11/27蛋白激酶响应渗透胁迫激发的Ca2+信号正调控胁迫应答(Li et al.,2025, Dev Cell)。鉴于BON1调控渗透胁迫下Ca2+信号,团队提出假说:BON1下游的CPK3/4/6/11/27激酶可能直接调控SNC1活性。通过遗传与生化分析,最终锁定CPK3为核心效应激酶:组成性激活形式的CPK3(constitutively active variant, CPK3CA)不仅能恢复渗透胁迫下bon1突变体的生长,还能抑制SNC1过表达导致的生长迟缓,而激酶失活突变的CPK3(K107M、ΔK107)则完全失效;机制解析表明,CPK3通过磷酸化TIR结构域的Ser26位点,抑制其NAD+水解酶(NADase)活性,从而阻断免疫信号过度激活,犹如为防御反应安装"刹车",巧妙控制防御强度,保障植物生长。
值得注意的是,这一机制在动物中同样保守:人类CAMK2D和TBK1激酶可磷酸化SARM1(包含TIR结构域蛋白)的对应位点(Ser567),抑制其NADase活性并阻断细胞死亡。这一平行机制证实TIR磷酸化是跨物种保守的‘免疫-生长’调控开关。该研究不仅为设计‘抗逆不减产’作物提供分子靶标(如编辑CPK3或SNC1磷酸化位点),还可能通过模拟TIR磷酸化机制,开发靶向SARM1的神经保护药物,为阿尔茨海默病等疾病提供新干预策略。
中国科学院分子植物科学卓越创新中心博士李俊(现就职于新疆省农科院)、陈思思和副研究员于波为该论文共同第一作者,赵杨研究员和万里研究员为共同通讯作者。博士后李庆忠、博士生刘睿佳以及博士后王再青参与了该研究。该项研究得到中国科学院基础与交叉前沿科研先导专项、中国科学院稳定支持基础研究领域青年团队、上海市科学技术委员会、植物性状形成与塑造全国重点实验室、植物高效碳汇重点实验室(中国科学院)以及中国科学院分子植物科学卓越创新中心逆境生物学研究中心的资助和支持。
论文链接:https://www.nature.com/articles/s41477-025-02012-x
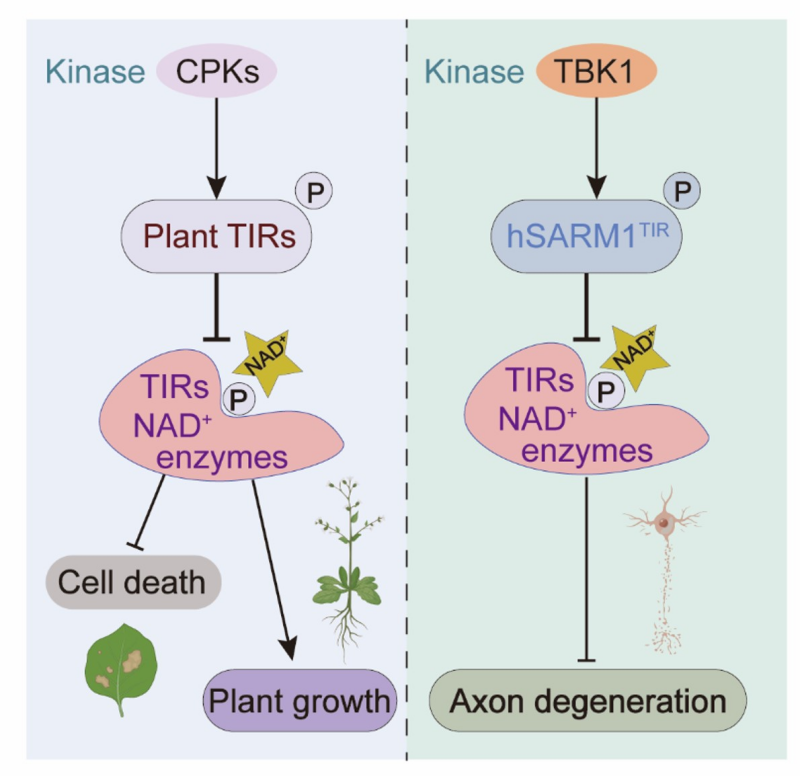
磷酸化修饰抑制TIR活性的保守分子机制
Phosphorylation modification inhibits TIR activity

Copyright © 2002-
中国科学院分子植物科学卓越创新中心 版权所有
地址:中国上海枫林路300号(200032)
电话:86-21-54924000
传真:86-21-54924015
Email: webmaster@cemps.ac.cn